 LOGIN/REGISTRATION
LOGIN/REGISTRATION
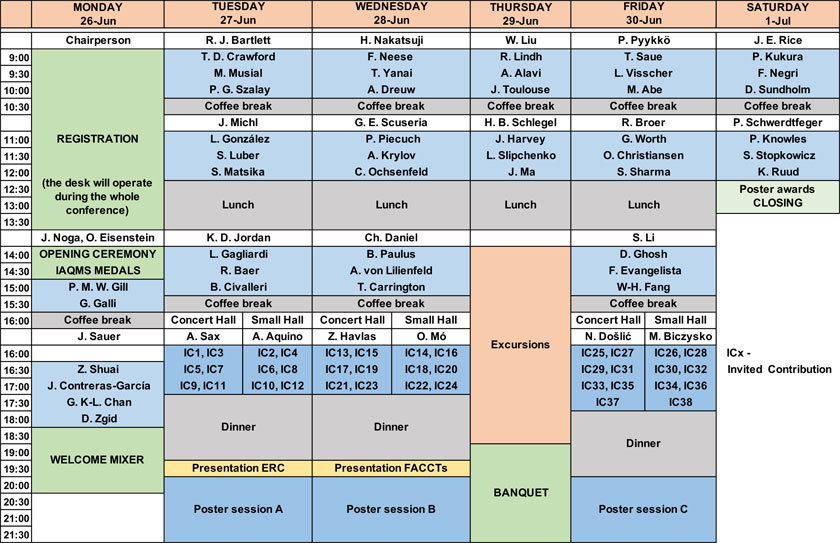
Programme Schedule
ICQC2023 conference HELPDESK is open during the entire conferece.
Molpro HELPDESK is open during coffee breaks on Tuesday and Wednesday afternoon.
FACCTs BOOTH SPACE is open during coffee breaks and poster sessions.
INVITED LECTURES
30 min lectures (including discussion)
- Minori Abe (Hiroshima University, Japan)
Relativistic quantum chemical calculations for uranium isotope fractionation in biotic and abiotic systems - Ali Alavi (Max Planck Institute for Solid State Research, Stuttgart, Germany)
Recent Progress with the Transcorrelated method: combining real-space approaches with quantum chemistry - Roi Baer (The Hebrew University of Jerusalem, Israel)
Stochastic Vector Techniques in Electronic Structure - Tucker Carrington (Queen’s University, Kingston, Canada)
Computing vibrational spectra of Van der Waals molecules when the harmonic approximation is useless - Garnet Chan (Caltech, Pasadena, USA)
The quantum chemistry of high temperature cuprate superconductors - Bartolomeo Civalleri (University of Turin, Italy)
Cost-effective hybrid HF/DFT composite methods for large-scale solid-state calculations - Julia Contreras-García (Sorbonne Universités, Paris, France)
From electron delocalization to predicting superconducting behavior - Daniel Crawford (Virginia Tech, USA)
Reduced-Scaling Coupled Cluster Theory in the Frequency and Time Domains - Ove Christiansen (Aarhus University, Denmark)
New quantum molecular dynamics methods: time-dependent vibrational coupled cluster theory with adaptive basis functions and direct dynamics approaches - Andreas Dreuw (Interdisciplinary Center for Scientific Computing, Heidelberg, Germany)
Algebraic diagrammatic construction schemes for molecular computations - Francesco Evangelista (Emory University, Atlanta, USA)
Molecular Electronic Structure at the Nexus of Classical and Quantum Computing - Weihai Fang (Beijing Normal University, China)
Variational quantum computation of molecular response properties on a superconducting quantum processor - Laura Gagliardi (University of Chicago, USA)
Localized Multireference Quantum Chemistry for Extended Systems - Giulia Galli (University of Chicago & Argonne National Laboratory, USA)
Electronic structure and coherent states of spin defects in molecules and solids - Debashree Ghosh (Indian Association for the Cultivation of Science, Kolkata, India)
Machine learning configuration space and matrix product state ansatz - Peter Gill (University of Sydney, Australia)
Economical Models for Electron Densities - Leticia González (University of Vienna, Austria)
Excited states and dynamics of coordination complexes - Jeremy Harvey (Katholieke Universiteit Leuven, Belgium)
Understanding the ‘Q’ of Biochemical Reactions: QM/MM Reaction Paths and Dynamics - Peter Knowles (Cardiff University, United Kingdom)
Coupling electrons, vibrations and photons in molecular quantum chemistry - Anna Krylov (University of Southern California, Los Angeles, USA)
Fun with X-rays: New theoretical developments in EOM-CC inspired by experimental light science - Philipp Kukura (University of Oxford, United Kingdom)
Polarisability and scattering at the single molecule limit - Anatole von Lilienfeld (University of Toronto, Canada)
Go EAST, young scientist – First principles view on chemical compound space - Roland Lindh (Uppsala University, Sweden)
The Three Billy Goats Gruff: Three Levels of Sophistication in SCF Orbital Optimization Procedures - Sandra Luber (University of Zurich, Switzerland)
Dynamic methods for computational spectroscopy and excited states in condensed phase - Jing Ma (Nanjing University, China)
Polarization Models for Charged and Polar Molecules - Spiridoula Matsika (Temple University, Philadelphia, USA)
Modeling experimental observables from excited state dynamics - Monika Musial (University of Silesia in Katowice, Poland)
High sectors of the intruder-free Fock space multireference coupled cluster method in the studies of excited states - Frank Neese (Max-Planck-Institut für Kohlenforschung, Mülheim, Germany)
On the opportunities offered by restricted open-shell methods - Fabrizia Negri (Università di Bologna, Italy)
Influence of diradical character and aggregation on optoelectronic properties of molecular materials: insights from cost-effective DFT-based calculations - Christian Ochsenfeld (Ludwig-Maximilians-Universität München, Germany)
Low-prefactor linear-scaling quantum-chemical methods - Beate Paulus (Freie Universität Berlin, Germany)
Computational Insight into Electrochemical Fluorination on a Nickel Anode - Piotr Piecuch (Michigan State University, East Lansing, USA)
Converging High-Level Coupled-Cluster Energetics via Semi-Stochastic, Selected-CI-Driven, and Adaptive CC(P;Q) Approaches - Kenneth Ruud (University of Tromsø, Norway)
Material properties from four-component relativistic DFT calculations - Trond Saue (Université Toulouse III-Paul Sabatier, France)
Does chemistry need more physics? - Sandeep Sharma (University of Colorado, USA)
Auxiliary field quantum Monte Carlo as a systematically improvable method for large systems - Zhigang Shuai (Tsinghua University, Beijing, China)
Time-dependent DMRG for electron dynamics in complex materials - Lyudmila Slipchenko (Purdue University, West Lafayette, USA)
Deciphering energy transfer in photosynthesis with polarizable embedding models - Stella Stopkowicz (Saarland University, Germany)
Advances and challenges in the study of the electronic structure of atoms and molecules in (strong) magnetic fields - Dage Sundholm (University of Helsinki, Finland)
Magnetically induced current-density susceptibilities and magnetic properties of molecules - Peter Szalay (ELTE Eötvös Loránd University, Budapest, Hungary)
Benchmarking aspects of ab initio fragment models for accurate excimer potential energy surfaces - Julien Toulouse (Sorbonne Universités, Paris, France)
Basis-set correction based on density-functional theory - Lucas Visscher (Vrije Universiteit Amsterdam, Netherlands)
Subsystem methods for density functional theory and quantum computing - Graham Worth (University College London, United Kingdom)
Benchmarking Non-adiabatic Quantum Dynamics Simulations Using the Molecular Tully Models - Takeshi Yanai (Nagoya University, Japan)
Analytic nuclear energy gradients of state averaged DMRG-CASSCF theory - Dominika Zgid (University of Michigan, Ann Arbor, USA)
Green’s functions for molecules and solids. What accuracy can we reach?
INVITED CONTRIBUTIONS
15 min lectures (including discussion)
- Tomas Bucko (Comenius University in Bratislava, Slovakia)
Accessing Free Energetics of the Adsorption Problem via Constrained Thermodynamic Integration in Internal Coordinates - Olga S. Bokareva (University of Kassel, Germany)
Optimally-tuned long-range corrected density functional for modeling excited stated of photoactive iron complexes - Hugh Burton (University of Oxford, United Kingdom)
Excited states, symmetry breaking, and multiple solutions in electronic structure theory - Bo Chen (Donostia International Physics Center, Donostia-San Sebastián, Spain)
The eXtreme Pressure-Polarizable Continuum Model (XP-PCM) and its applications to pericyclic reactions under pressure - Ji Chen (Peking University, China)
Electronic structure of molecules and solids with neural network quantum Monte Carlo - Pavlo O. Dral (Xiamen University, China)
Accelerating and improving quantum chemistry and dynamics with artificial intelligence - Achintya Kumar Dutta (IIT Bombay, India)
Reduced cost four component relativistic coupled cluster method based on natural spinors - Alessandro Genoni (CNRS & University of Lorraine, Metz, France)
X-ray Restrained Wavefunction Approach: A Quantum Crystallographic Tool to Extract Exchange-Correlation Potentials from X-ray Diffraction Data? - William Glover (NYU Shanghai, China)
Conical intersections in solution with polarizable embedding - Andreas Hansen (Universität Bonn, Germany)
Beyond the Gold Standard: Sub-kcal/mol Accuracy for Isomerization and Conformational Energies of Larger Molecules from Explicitly Correlated Local Coupled Cluster Methods - Erik Donovan Hedegård (Southern Denmark, Odense, Denmark)
Treating transition metals in solvents and proteins properly - Denis Jacquemin (Université CNRS CEISAM, France)
Ultra-accurate transition energies with EOM-CC4 - Thomas Jagau (KU Leuven, Belgium)
Molecular Auger spectra from complex-scaled coupled-cluster theory - Zsuzsanna Koczor-Benda (University of Warwick, United Kingdom)
Computational Molecular Design for Terahertz Detection and Surface-Enhanced Applications - Agnieszka Krzemińska-Kowalska (Lodz University of Technology, Poland)
Efficient computations and visualization of dispersion energy for multireference systems using Cholesky decomposition, with a focus on analyzing excited-state interactions - Mitchell Lahm (University of Georgia, Athens, United States)
Concordant Mode Approach for Molecular Vibrations - Stephanie Lambie (University of Auckland, New Zealand)
Metallic crystal growth within a liquid gallium solvent - Örs Legeza (Wigner Research Centre for Physics, Budapest, Hungary)
Predicting the FCI energy of large systems to chemical accuracy from restricted active space density matrix renormalization group calculations - WanZhen Liang (Xiamen University, China)
Exploring Molecular Photophysical and Photochemical Properties Using Linear Response Time-Dependent Density Functional Theory with Classical Embedding - Hans Lischka (Texas Tech University, Lubbock, USA)
The Assessment of the Polyradical Character of Polycyclic Aromatic Hydrocarbons by Means of Multireference and Density Functional Theory Methods - Rabindranath Lo (Czech Academy of Sciences, Prague, Czech Republic)
Enhanced Stability of Covalent Dative Bond in the Presence of a Solvent - Alessandro Lunghi (Trinity College, Dublin, Ireland)
Towards exact first-principles predictions of spin-phonon relaxation - Haibo Ma (Shandong University, Qingdao, China)
New Density Matrix Renormalization Group Approaches for Strongly Correlated Systems Coupled with Large Environments - Edit Matyus (Eötvös Loránd University, Budapest, Hungary)
Towards a relativistic QED for atomic and molecular bound states - Torsha Moitra (Hylleraas Centre for Quantum Molecular Science, UiT The Arctic University of Norway)
Ab-initio attosecond pump-probe transient absorption spectroscopy with relativistic TDDFT - Jógvan Magnus Haugaard Olsen (Technical University of Denmark DTU, Kongens Lyngby, Denmark)
MiMiC: A High-Performance Framework for Multiscale Modeling in Computational Chemistry - Ángel Martín Pendás (Universidad de Oviedo, Spain)
Persistence of atoms in molecules: from wavefunctions to open quantum systems - Juan Peralta (Central Michigan University, Mount Pleasant, USA)
Removing Self-interaction Error from Density Functional Theory Calculations with Fermi-Löwdin Orbitals - Jiri Pittner (J. Heyrovský Institute of Physical Chemistry ASCR, Prague, Czech Republic)
Hilbert space multireference coupled clusters tailored by matrix product states - Dennis Salahub (University of Calgary, Canada)
Towards ML-Accelerated Discovery of Nanocatalytic Materials and Mechanisms - Christian Schilling (Ludwig Maximilian University, Munich, Germany)
Density Matrix Functional Theory for Excited States - Dumitru-Claudiu Sergentu (Univ Rennes CNRS ISCR, Rennes, France)
The resolution of the weak-exchange limit made rigorous, simple and general in binuclear complexes: A theoretical feat - Petr Slavíček (University of Chemistry and Technology, Prague, Czech Republic)
Activating UV chromophores with red light: Proton-coupled energy transfer - Michal Tomza (University of Warsaw,Poland)
Quantum resonances in ultracold atom-ion and atom-molecule collisions - Claire Tonnelé (Donostia International Physics Center Euskadi, Spain)
Rationalization and tuning of doublet emission in organic radicals - Takashi Tsuchimochi (Kobe University, Hyogo, Japan)
Recent developments of quantum algorithms for excited states - Demeter Tzeli (University of Athens, Greece)
Electronic structure and bonding properties of iron-sulfur model complexes - Oscar Ventura (Universidad de la República, Montevideo, Uruguay)
Validation and application of the SVECV-f12 composite method for reaction barriers and heats of formation